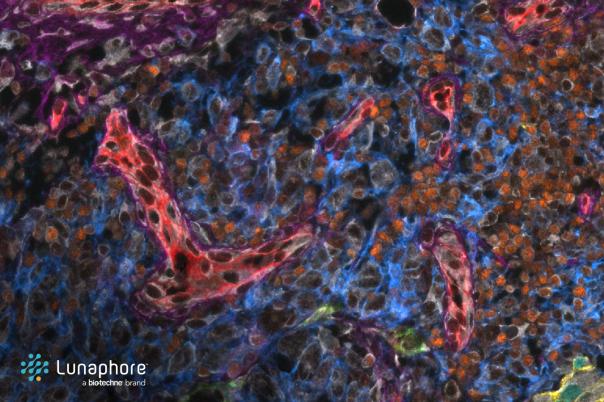
Kieran Campbell, Assistant Professor & Group Leader, University of Toronto & Lunenfeld-Tanenbaum Research Institute has developed three key tools to assist with the analysis and interpretation of spatial genomics data. He also highlighted how to design antibody panels using single-cell RNA sequencing atlases and how to identify cell phenotypes from spatial data when there are multiple segmentation errors.
Having struggled to find software that met all of his needs, Campbell decided to develop his own spatial software. The first tool he introduced is called Rakaia, a browser-based tool for spatial biology analysis. Rakaia integrates large spatial biology data sets and performs a variety of functions such as interactive analysis, visualisation, and annotation of these sets. Then, one can export the analysis to a shareable HTML report where one can view spatial data and H&E side by side to assist with annotation.
Secondly, he developed antibody panels for a multiplexed imaging analysis experiment. His goal was to use single-cell RNA sequencing to guide antibody panel design to find novel targets and markers. Cytomarker is a web app designed for interactive antibody panel design using single-cell RNA sequencing data. It helps in identifying novel markers and capturing rare cell populations, enhancing the design of antibody panels for spatial experiments. The app offers many different ways to visualise data and the panel quality score feature quantitively shows how well one is capturing the variation in the data set.
Campbell carried out a perfect concept experiment using Cytomarker where they conducted single-cell RNA sequencing on a healthy human breast. His team quantified single-cell populations in the luminal and basal epithelial cells, fibroblasts, endothelial cells and immune cells. Ultimately, the platform identified canonical and novel markers that were validated through imaging mass cytometry.
Next, Campbell introduced STARLING, a probabilistic method for identifying cell phenotypes in the presence of segmentation errors. This tool improves the accuracy of cell type identification by accounting for segmentation inaccuracies in spatial data. The team also introduced a “plausibility score” based on known protein co-expression rules. A comparative study with traditional methods showed that STARLING demonstrated improved clustering fidelity.
Finally, Campbell summarised by encouraging the audience to access STARLING on Google or GitHub. He also mentioned that Cytomarker is an open-source tool that is free for anyone to use. The widespread availability and use of these tools promote collaboration.