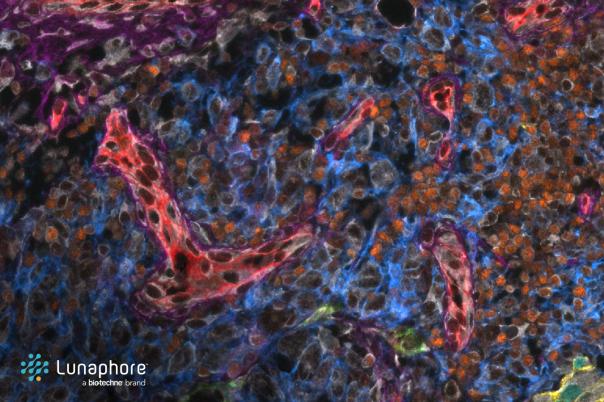
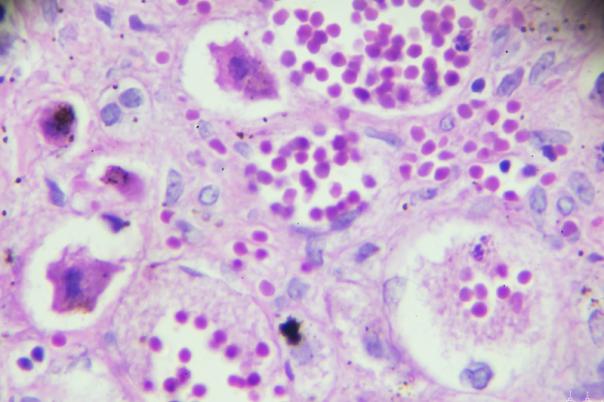
Cellular networks play an important role in tissue homeostasis and disease, especially in tumours. There are many ways in which cells can exchange molecules and information, but the primary method is via ligand-receptor interactions. Jacques Collange’s talk focuses on unravelling this complex network of interactions when studying disease using models of cellular networks and single cell spatial data.
Using single cell transcriptomics, data can be clustered so different cell populations can be recognised. From here, ligand-receptor interactions can be inferred based on observed expression in the cell population and by consulting a database of known interactions. If the database considers signalling downstream of the receptor, intracellular networks can be drawn from these data too.
Collange then considered how to use this methodology applied to bulk data. This is trickier, because all the cell types are mixed in one measure: the transcriptome or proteome. There are still plenty of interesting data to glean from bulk datasets – Collange said that this kind of analysis can still be performed by making the statistical models more sophisticated to deal with less accurate data.
Collange and his team have produced the BulkSignalR model to infer ligand-receptor cellular networks from bulk and spatial transcriptomic datasets. The model includes downstream signalling and the target gene regulation, forming a complete pattern observable through correlation analysis. This outputs tables of interactions with estimated statistical significance.
The framework integrates downstream signalling and target gene regulation, allowing prediction of intracellular activities at the target cell population. Applying this to Visium data reveals spatial specific activity of pathways triggered by ligand receptor interactions, showing meaningful spatial patterns typical of cancer cells and stroma.
This approach provides insights into cellular interactions and spatial patterns in single cell data. With advancements in instrumentation, single cell proteomics is expected to emerge strongly, providing comparable insights to transcriptomics in the coming years.