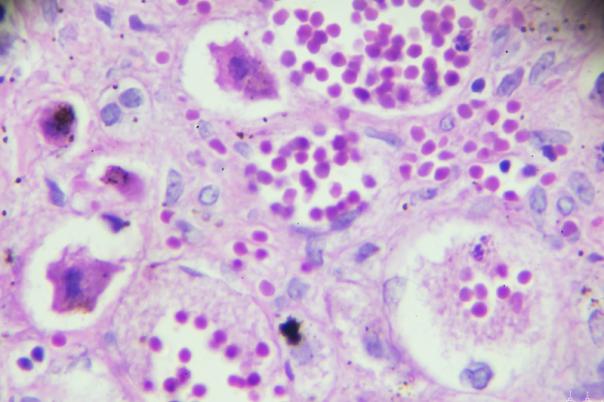
Human organs include a diverse array of cell types which each have their own function. This cell type function influences tissue function which in turn influences organ function. Bulk omics technologies have been the foundation for examining this cell function. Single cell technologies have taken this further by isolating and profiling individual cells’ transcriptome which can reveal developmental trajectories and cellular changes, particularly in relation to diseases like cancer.
This gave rise to the field of interceptive medicine: quantifying the early events that lead to pathology and trying to stop them, rather than waiting until the disease is onset and try to cure tissues that are already damaged. Perturbation modelling is one way to do this; it applies screening techniques to biological systems and uses AI modelling to reconstruct the early stages that led to the tissue trajectory.
Ishaque then went on to explain how this concept was used in his teams work on preeclampsia, a significant pregnancy-related condition with limited therapeutic options. His team aims to leverage single cell technologies to improve understanding and treatment options.
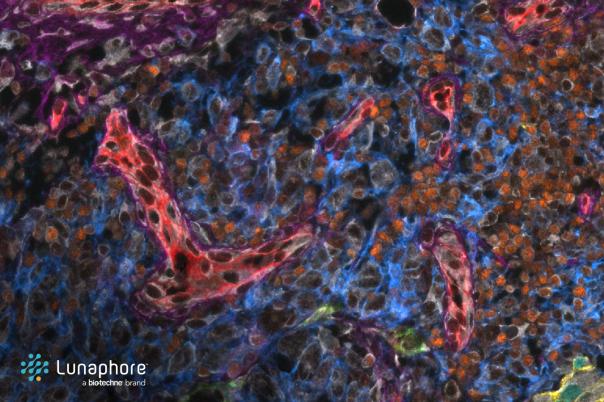
The presentation also highlights the potential of spatial transcriptomic technologies. This allows researchers to understand not only the single cell gene expression information, but also the spatial organisation of what has been measured. Ishaque explained the directions that the up-and-coming field is heading in, including an increase in resolution, implementation of three-dimensionality, and multiomic methods.
One exciting development is the potential for AI models to predict molecular features directly from histology images, bypassing intermediate steps. Proof-of-principle experiments have shown that this is possible, and efforts are underway to scale this up across different spatial transcriptomics technologies.
The concept of overlapping cells in supposedly 2D spatial models can be one of the main challenges in spatial transcriptomics. It can lead to confusing signals and inaccurate data interpretation. In response, Ishaque’s team developed a tool called Overl.py, this unsupervised method generates maps that highlight areas of high overlap, improving the accuracy of spatial gene expression analysis.